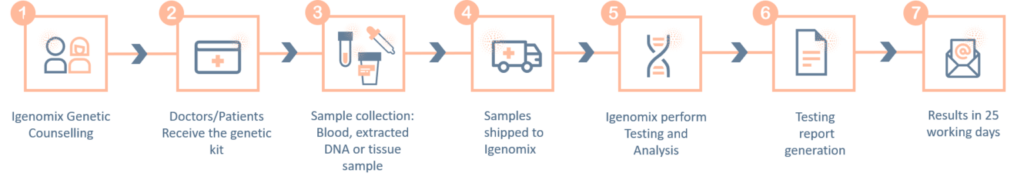
Panel de precisión del síndrome de Ehlers-Danlos
Los síndromes de Ehlers-Danlos (EDS) son un grupo heterogéneo clínica y genéticamente de trastornos del tejido conectivo, donde el defecto genético afecta la síntesis y estructura del colágeno y del tejido conectivo. Se caracteriza por hipermovilidad, fragilidad cutánea e hiperextensibilidad.