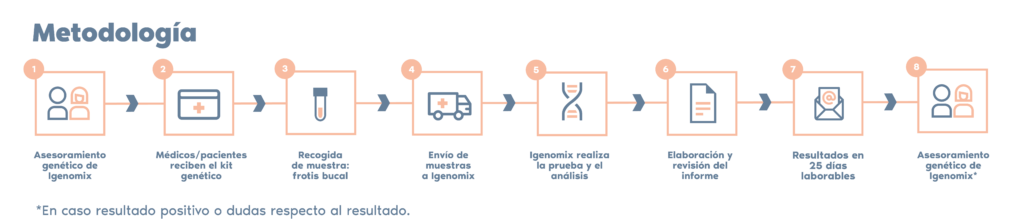
Panel integral de precisión de cardiología
La miocardiopatía es un grupo de afecciones con un fuerte trasfondo genético que dificultan estructuralmente que el corazón bombee sangre al resto del cuerpo debido a la debilidad de los músculos del corazón.